

Studie opzetten
DiaPregNL Cohort
Samenvatting verantwoordelijkheden van deelnemende centra:
- Communicatie met coördinerend team bij vragen of knelpunten. Deelname aan feedbackrondes en pilotmeetings.
- Regelen van lokale ethische goedkeuring en ondertekening van de CTA.
- Lokale werving van deelnemers. Veilig bewaren van ondertekende toestemmingsformulieren (PIFs). Bijhouden van een actueel ISF, inclusief een beveiligd lokaal sleutelbestand.
- Data verzamelen door het invoeren van gegevens in Castor eCRF en het verzenden van vragenlijsten. Bewaken van datakwaliteit en volledigheid binnen eigen centrum.
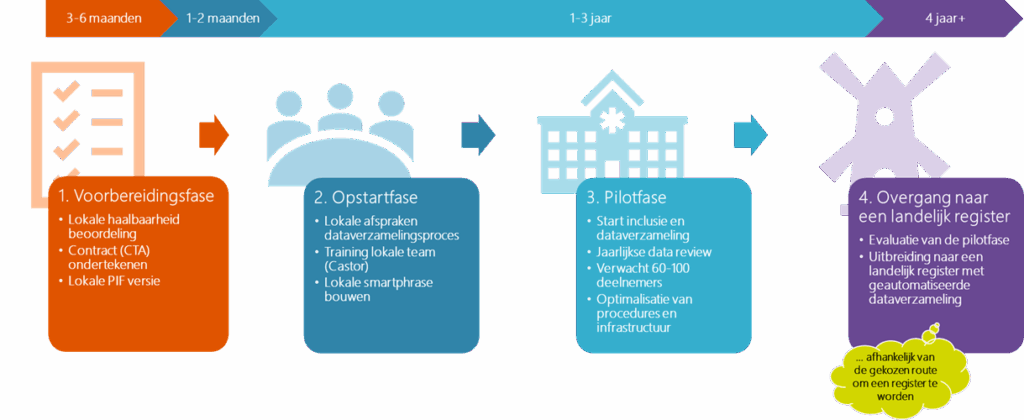
Hieronder vind je een uitgebreide uitleg van de belangrijkste stappen in elke fase.
Voorbereidingsfase
Opstartfase
Wat houdt deze fase in?
De opstartfase begint zodra de lokale goedkeuring is verkregen en het contract (CTA) door beide partijen is ondertekend.
Tijdens het opstartoverleg met de coördinerend onderzoeker worden alle praktische afspraken voor de uitvoering van het onderzoek besproken, zoals de werkwijze voor dataverzameling, de onderzoeksprocedure en de inrichting van het ISF. Ook worden afspraken gemaakt over de fysieke opslag van de PIF’s. Daarnaast krijgt het lokale team training in het gebruik van Castor, en wordt een lokale smartphrase opgesteld zodat standaardteksten in het patiëntendossier eenvoudig en consistent kunnen worden gebruikt.
Castor eCRF
In deze studie gebruiken we Castor electronic Case Report Form (eCRF), een veilig online platform voor het verzamelen en beheren van onderzoeksgegevens. Het is gebruiksvriendelijk, waardoor training niet nodig is. Vanwege de gevoelige gegevens is het belangrijk om twee-factor-authenticatie (2FA) in te schakelen; zonder 2FA heb je geen toegang tot de studieomgeving. Tijdens de opstartmeeting met de coördinerend onderzoeker wordt een korte demo gegeven over het correct invullen van de eCRF.
Elk centrum krijgt toegang tot twee locaties binnen de studieomgeving: hun eigen ziekenhuis, waar ze echte patiëntgegevens invoeren, en een testlocatie, die gebruikt kan worden om formulieren en datainvoer te oefenen. Hoewel de eCRF grondig is getest, kunnen er tijdens het invoeren van gegevens nog fouten optreden. Deze dienen te worden doorgegeven aan de coördinerend onderzoeker. Data wordt verzameld voor onderzoek in twee aparte studieomgevingen:
- DiaPregNL Register: bevat gepseudonimiseerde onderzoeksdata uit de deelnemende centra, bijvoorbeeld over diabeteszorg tijdens de zwangerschap.
- Koppel DiaPregNL: bevat persoonlijke data van deelnemers die expliciet toestemming hebben gegeven, gescheiden van de data in het DiaPregNL Register.
Deelnemers moeten toestemming geven voor het gebruik van hun gegevens, die strikt beveiligd worden opgeslagen en beheerd, volgens de AVG-richtlijnen. De gegevens worden verzameld door het interne geneeskunde-team. We vragen van pilot centra om feedback te geven die we kunnen gebruiken om het systeem te verbeteren voordat het nationale register wordt gelanceerd, wat binnen drie jaar gepland is.
Investigator Site File (ISF)
Elke site dient een lokale ISF bij te houden, bij voorkeur als een map op een gedeelte, beveiligde netwerk. Dit map bevat het Lokale Sleutelbestand, met de persoonlijke data van deelnemers, en alle relevante studiedocumentatie (zoals het protocol, PIFs, en ondertekende CTA). Het bestand dient veilig te worden opgeslagen en is uitsluitend toegankelijk voor bevoegde medewerkers.
Fysieke Opslag van PIF’s
De deelnemende ziekenhuizen dienen gedurende de pilotfase fysieke versie van de ondertekende patiënteninformatiebrief en toestemmingformulier veilig te bewaren.
Na de pilotfase kunnen de PIF’s mogelijk naar Amsterdam UMC worden overgebracht, maar het langetermijnplan voor opslag is nog in ontwikkeling en wordt later gecommuniceerd.
Smartphrase
Tijdens standaard poli afspraken kan een smartphrase worden gebruikt om de gegevens die nodig zijn voor het onderzoek consistent te noteren. Dit bespaart tijd, omdat alle benodigde informatie op één plek staat in de notities, zolang de smartphrase volledig ingevuld wordt tijdens het patiëntcontact.
Hoewel de gegevens nog handmatig naar Castor overgedragen moeten worden, zorgt de smartphrase ervoor dat alle relevante informatie makkelijk toegankelijk is en het proces van gegevensinvoer sneller en efficiënter verloopt.
Dataverzameling volgens AVG
Wij hanteren strikt de richtlijnen van de Algemene Verordening Gegevensbescherming (AVG). We vragen elke deelnemer toestemming om hun gegevens te verzamelen en te delen met Amsterdam UMC voor het DiaPregNL Register, en we vragen een aparte toestemming voor het Koppel DiaPregNL.
We hebben een positief privacyadvies gekregen van de Functionaris Gegevensbescherming van Amsterdam UMC. Voor een overzicht van hoe de data binnen de studie stroomt, zie Figuur hieronder.
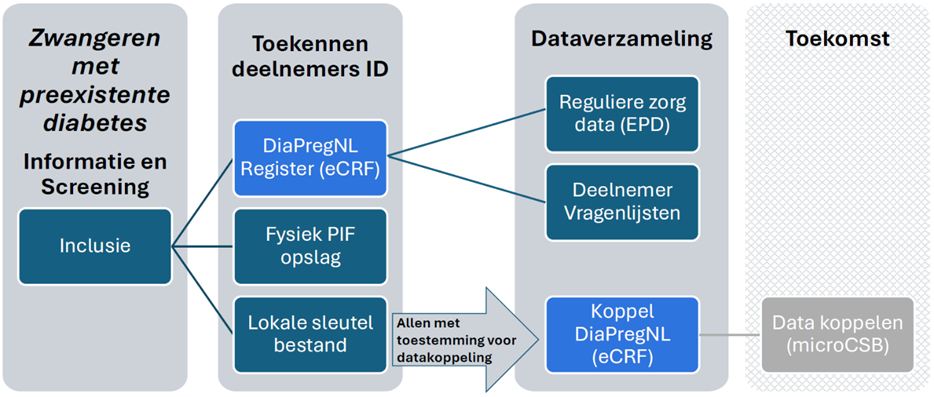
- Beveiliging bij de lokale centra: Een beveiligde locatie binnen het lokale computernetwerk is verplicht voor het ISF, dat gedeeld wordt met de coördinerend onderzoeker en het lokale onderzoeksteam. Het lokale sleutelbestand moet extra beveiligd worden (bijvoorbeeld met een wachtwoord en 2 Factor Authenticatie). Er dient een afgesloten kast beschikbaar te zijn voor de opslag van de ondertekende PIF’s.
- Pseudonimisering: De onderzoeksdata in het DiaPregNL Register worden zodanig verwerkt dat de identiteit van de deelnemers niet direct herleidbaar is zonder aanvullende informatie, welke apart wordt opgeslagen in het lokale sleutelbestand en in de Koppel DiaPregNL (voor deelnemers die toestemming hebben gegeven voor datakoppeling en delen met het coördinerend centrum).
- Beveiliging bij het coördinerend centrum: Beide registers worden beheerd in Castor eCRF en voldoen aan strikte beveiligingsprotocollen Alleen het coördinerende team en het lokale team kunnen de deelnemers van hun eigen ziekenhuis zien. Elk ziekenhuis ziet alleen zijn eigen deelnemers.. Binnen Amsterdam UMC is voor Castor geen DPIA vereist. Mocht de lokale haalbaarheidscommissie toch een DPIA vereisen, kunnen we samenwerken om deze op te zetten voor de lokale situatie.
Pilotfase
Wat houdt deze fase in?
De pilotfase start na het opstartoverleg met de coördinerend onderzoeker.
In deze fase begint de werving van deelnemers en de dataverzameling op de locatie. De voortgang en eerste ervaringen met het onderzoeksproces worden geëvalueerd na 3, 6 en 12 maanden, en er vindt jaarlijks een datareview plaats om de kwaliteit en volledigheid van de gegevens te beoordelen. Op basis van de evaluaties worden de procedures en infrastructuur waar nodig verder geoptimaliseerd.
Deze fase zal doorgaan totdat het onderzoek uitgroeit tot een officieel register, naar verwachting rond 2027. Meer over de stappen om een register te worden, is in de volgende sectie.
Werving en Inclusiecriteria
De werving vindt hoofdzakelijk plaats via de interne geneeskunde, maar gynaecologen kunnen ook patiënten informeren over het onderzoek. We hebben een website en een eenvoudige flyer die in de spreekkamers kan worden opgehangen.
We adviseren om mogelijke deelnemers tijdens de eerste afspraak in het eerste trimester toe te lichten en ze na de termijnecho te includeren. Inclusie op een later moment is mogelijk maar leidt tot minder complete data.
Inclusiecriteria
- Zwanger, in eerste trimester
- Volwassen (18+), wilsbekwaam
- DM diagnose voor zwangerschap
- Spreekt Nederlands of Engels*
*We hopen binnenkort PIF’s en vragenlijsten in meer talen beschikbaar te hebben.
Dataverzameling proces
De dataverzameling in de pilotstudie vindt plaats tijdens reguliere zorgcontacten met de deelnemende patiënten, zonder dat er extra studiebezoeken nodig zijn. Er zijn vier meetmomenten: één per trimester van de zwangerschap en één postpartum (Figuur 2). Alle verzamelde gegevens worden ingevoerd in het Castor eCRF.

Voordat we data automatisch kunnen verzamelen, hebben we een kortetermijnoplossing ontwikkeld. Om het invoeren van data te vereenvoudigen, is er een voorbeeld smartphrase gemaakt voor alle meetmomenten. Deze kan worden gebruikt tijdens standaardafspraken in de zwangerschap. Zo worden de basisgegevens die nodig zijn voor het register al tijdens de reguliere consulten verzameld en later in het DiaPregNL Register ingevoerd.
Diabetesbeheer
Het meest van de verzamelde data betreft diabetesbeheer en wordt verzameld door het team van interne geneeskunde tijdens reguliere spreekuren. Hierbij gaat het om informatie over glucosemonitoring, insuline(pomp)gebruik, medicatie en CGM-data (indien beschikbaar). De data worden viermaal verzameld: eenmaal per trimester en eenmaal postpartum.
Zwangerschapsuitkomsten
Momenteel vragen we het interne geneeskundeteam om de obstetrische voorgeschiedenis en zwangerschapscomplicaties in te vullen op basis van het dossier en de patiënte anamnese. We zijn ons ervan bewust dat dit niet de meest betrouwbare bron is en hopen voor het register data te verkrijgen via Perined of rechtstreeks uit het ziekenhuisinformatiesysteem. Als een ziekenhuis nauwer samenwerkt met het gynaecologieteam en het haalbaar is om de vragen door gynaecologen te laten invullen, bespreken we dit graag.
Afsluiten van een deelnemer
Elke deelnemer in de studie krijgt een status toegewezen wanneer de deelname wordt beëindigd, hetzij door voltooiing van de dataverzameling, hetzij door voortijdig stoppen of verlies van follow-up (zie Tabel hieronder). Het is belangrijk om de reden van afsluiting nauwkeurig vast te leggen in het Castor onderzoeksomgeving.
Voor elke afgesloten deelnemer wordt de datum van afsluiting vastgelegd. Indien mogelijk wordt aanvullende informatie over de reden voor afsluiting verzameld, met respect voor privacy en ethische richtlijnen. Deze gegevens worden gebruikt voor datakwaliteitscontrole, analyses van uitval en het correct interpreteren van de resultaten van de cohortstudie.

Gebruik van onderzoeksgegevens
Op dit moment mag alleen Amsterdam UMC van de verzamelde onderzoekgegevens gebruikmaken. De deelnemende ziekenhuizen kunnen gebruik maken van de eigen verzamelde onderzoeksgegevens, zoals vastgelegd in de Clinical Trial Agreement. In de PIF is al opgenomen dat gegevens in de toekomst mogelijk gedeeld kunnen worden met derde partijen, ook buiten de EER, maar dat is op dit moment nog niet van toepassing. Dergelijke datadeling zal pas mogelijk zijn zodra het project formeel wordt omgezet in een register. In dat geval zal er een aparte Joint Research Data Agreement worden opgesteld, die door alle deelnemende centra moet worden ondertekend. Onderdeel daarvan zal ook een toetsingscommissie zijn die aanvragen voor secundair gebruik van de onderzoeksgegevens beoordeelt. Eventuele wijzigingen in het gebruik of de structuur van het register zullen altijd vooraf worden afgestemd met en gecommuniceerd aan de lokale hoofdonderzoekers.